
As part of a series on how the prognosis and treatment for different diseases have changed over time, here are the basics of cystic fibrosis (CF) – as well as information on new therapies coming on stream, and what the future holds for those with CF.
What is it?
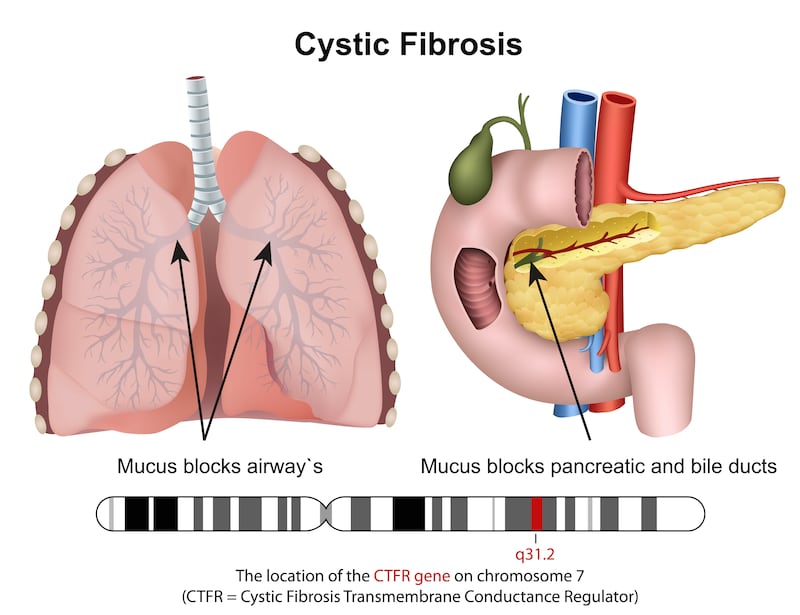
Cystic fibrosis is a rare genetic condition caused by up to 2,000 known mutations in genes inherited from both parents. These genetic mutations affect the body’s ability to make and direct the so-called CFTR (cystic fibrosis transmembrane conductance regulator) protein which helps salt and water move in and out of the body’s cells. The reduced amount of this protein leads to the build-up of thick sticky mucus in the body organs (especially in the lungs and digestive system) which can result in life-threatening lung infections.
Who gets it?
Ireland has the highest rate of CF in the world with about 1,400 people living with CF here at any one time. One in 25 people in Ireland carry a CF gene, but for someone to be born with CF, both parents must carry the faulty gene. If both parents have the gene, there is a one in four chance that the child will have CF. About 33 new cases of CF are diagnosed each year through the newborn screening programme which was introduced into Ireland in 2011.
What are the symptoms?
The main symptoms of CF start in early childhood. They include persistent coughing, wheezing and shortness of breath, due to the build-up of mucus in the lungs. When the mucus becomes infected with bacteria, this causes respiratory infections including pneumonia and bronchitis. Many children with CF have difficulty gaining weight, in spite of a good appetite, due to disruption of the natural enzymes which help break down food. The condition also results in difficulty with bowel movements.
READ MORE




What are the treatments?
People with CF must manage their condition on a daily basis. This includes using inhalers to open up the airways to improve breathing, using nebulisers with medicines to break down mucus and taking antibiotics to prevent and/or treat chest infections. Daily chest physiotherapy and sinus rinses also help to clear the perpetual build-up of mucus.
Because the condition disrupts the digestive system, those with CF need to ensure that they eat a high-calorie diet so that they get enough nutrients and vitamins. Enzymes are also advised to aid digestion and laxatives to increase bowel movements to prevent constipation.
What are the services like?
One of the biggest developments in the treatment of CF in Ireland was the establishment of specialised multidisciplinary CF outpatient and inpatient centres at some hospitals in Ireland during the 2010s. At these dedicated paediatric and adult CF centres, people with CF gain access to medical consultants, CF clinical nurse specialists, dieticians and physiotherapists. This access also means that individuals with CF with lung infections can be admitted to hospital quickly if necessary and given intravenous antibiotics in single rooms so they don’t become re-infected while there.
What new therapies are coming on stream?
The introduction of new drug therapies such as Kalydeco, Orkambi, Symkevi and Kaftrio in the last decade has given people with CF a better quality of life and increased longevity. These so-called CFTR modulators treat the underlying cause (ie the protein defects that result from the gene mutation) of CF. However, gaining access to these new drugs for specific age groups has often come about after long-fought battles with the HSE and media campaigns led by Cystic Fibrosis Ireland.

Speaking in a video recorded to celebrate the 60th anniversary of Cystic Fibrosis Ireland, CF ambassador Keith McCabe said, “There should not be anyone dying of CF in Ireland now [due to lack of treatment]. Everyone should have access to CF drugs. There shouldn’t be any minority groups excluded because of money.”
What other issues are there?
Many people with CF have some form of glucose intolerance which means that they have to monitor their glucose levels and follow a well-balanced diet.
As people with CF are living longer, many experience declines in their lung function which result in the need for lung transplants. Access to isolated rooms for people with CF undergoing surgery and recovery from transplants remains of crucial importance to prevent infection at a time when their immunity system is further compromised.
People with CF who want to start a family may need fertility treatments to aid conception and Cystic Fibrosis Ireland is currently campaigning for publically funded IVF treatment for people with CF who want to have children.
How does the experience of someone with CF now compare to that of 50 years ago?
In the aforementioned video, Philip Watt, the chief executive of Cystic Fibrosis Ireland, said that for people with CF to reach secondary school in the 1960s was seen as an achievement. Prof Muiris Fitzgerald, emeritus professor of medicine at University College Dublin, and the first CF adult consultant in Ireland, said that at that time CF patients were malnourished and had very bad lung disease.

“Very few people survived beyond their mid-20s. As a team and in the CF community, we used to celebrate the first wedding, the first baby, the first university degree, as these were incredible milestones for people with CF then.”
Now, about six in 10 people with CF in Ireland are over 18 and many people with CF live until their 50s. The number of adults with CF in 2023 is three times higher than 10 years ago. In 2021, a study found that the average age of survival is rising by one year every year and now stands at 52 for those born with CF between 2017 and 2021.
Colm Priestley (36), who has CF and is a father of two children, says that while CF has a massive treatment burden and he is on a lot of medications for his lungs and digestive system, his lung function has never been better. “All my life, I’d have generally considered that I was in the last five to 10 years of my life. But, we are now at a point with medication where we don’t have that limit. I’m not putting that limit on myself anymore and I hope others can do the same.”
What does the future hold?
Prof Paul McNally, respiratory consultant at Children’s Hospital Ireland at Crumlin says the treatments for CF have been steadily getting better and better at slowing down the decline in lung function and nutrition.
“The issues for people with CF now are health promotion and preventing ill health rather than waiting to treat complications when they arise,” says Dr McNally. He adds the pace of research and development of new innovative therapies for CF means that breakthroughs continue to occur. “It is not out of the question to suggest that in my working life a cure for CF may be found.”










